C1CN1
C#CCC#CC
CN1C=NC2=C1C(=O)N(C(=O)N2C)
Format: [SMILE] (one per line)
Welcome to ELAYA SMILES
Energy-minimized Linear-to-structure Atom Yielding Algorithm
A web platform that converts SMILES notation into optimized 3D molecular geometries — interactively and for free.
What is a SMILES string?
SMILES — Simplified Molecular Input Line Entry System — is a compact text-based notation for encoding molecular structure. Each character encodes atoms, bonds, branches, rings, and stereochemistry.
It is the standard exchange format in databases such as PubChem, ChEMBL, and ZINC — the starting point for any QM/MM or docking pipeline.
| Molecule | SMILES | Notes |
|---|---|---|
| Benzene | c1ccccc1 | Aromatic ring |
| Dopamine | C1=CC(=C(C=C1CCN)O)O | Catecholamine |
| Serotonin | C1=CC2=C(C=C1O)NC=C2CCN | Indole scaffold |
| Glucose | OC[C@H]1O[C@@H](O)… | Stereocenters (@) |
| Oxytocin | CC(C)C[C@H](NC(=O)… | Nonapeptide |
C / c= #( )1…9@ @@[NH3+]/ \Why does 3D geometry matter?
A SMILES string describes how atoms are connected — but chemistry happens in three dimensions. Biological activity, receptor binding, spectroscopic signatures, and reaction pathways are all governed by the precise spatial arrangement of atoms.
Generating an accurate 3D structure is often a critical first step before molecular dynamics simulations, docking studies, quantum-chemical calculations, or certain machine-learning models that rely on structural molecular features.
Three algorithms, one interface
ELAYA offers three complementary 3D construction methods, each suited to different use cases.
Distance-geometry embedding (ETKDGv3) followed by force-field minimization.
Template-based coordinate builder. Offers broader chemical coverage than many toolkits, though complex systems such as organometallics may still require specialized methods for accurate geometries.
Graph-theoretic spring layout. Fast and lightweight, useful for topological exploration.
RDKit — ETKDGv3 pipeline
The most precise method implemented in ELAYA
Chem.MolFromSmiles(smiles)
Builds the molecular graph: nodes = atoms, edges = bonds. Validates SMILES syntax.
Chem.AddHs(mol)
Adds all hydrogens explicitly — required for correct 3D geometry.
AllChem.EmbedMolecule(mol, ETKDGv3())
Experimental Torsion Knowledge Distance Geometry v3 (ETKDGv3). Incorporates knowledge-based torsional preferences derived from experimental and computational data to generate realistic 3D conformations. A fixed random seed (665) is used for reproducibility.
UFFOptimizeMolecule / MMFFMinimize
Minimizes energy with UFF or MMFF94 (max 2000 iterations). If MMFF94 fails, falls back to UFF automatically.
→ XYZ + MOL block
Generates the XYZ file (N atoms, SMILES comment, coordinates) and a MOL block for the 3Dmol.js viewer.
Force field optimization
A set of mathematical equations and parameters that model forces between atoms: bond stretching, angle bending, dihedral torsion, and non-covalent interactions (van der Waals, electrostatics). Minimizing energy with these equations produces more realistic 3D geometries than a bare embedding.
General — covers the entire periodic table. Based on atomic hybrid geometries.
Parametrized for drug-like organic molecules. High precision in bond geometry and angles.
Conformational search with GLOMOS
GLOMOS (Genetic-algorithm-based Local Optimization of MOlecular Structures) implements a heuristic genetic algorithm to explore the conformational space of rotatable bonds using ANI-based neural network potentials. It is integrated as a streaming endpoint (/api/convert/stream) with real-time Server-Sent Events (SSE) feedback.
Most molecules exist as a family of interconverting 3D shapes — conformers — differing in the rotation of single bonds. GLOMOS aims to identify low-energy conformers with accuracy approaching quantum-chemical methods for many organic systems, while avoiding the high computational cost of DFT calculations.
| Parameter | Default | Description |
|---|---|---|
initpop | 4 | Initial population size |
matings | 2 | Crossovers per generation |
mutants | 2 | Random mutations per generation |
generations | 3 | Total number of generations |
energy_cutoff | 4.0 | Energy window (kcal/mol) |
tol_similarity | 0.95 | Similarity tolerance between conformers |
cutoff_population | 8 | Max population size |
nof_repeats | 2 | Repeats before stopping |
nof_stagnant | 3 | Stagnant generations → stop |
ani_model | ANI1ccx | Neural potential (ANI1x / ANI1ccx / ANI2x) |
Genetic algorithm pipeline & real-time streaming
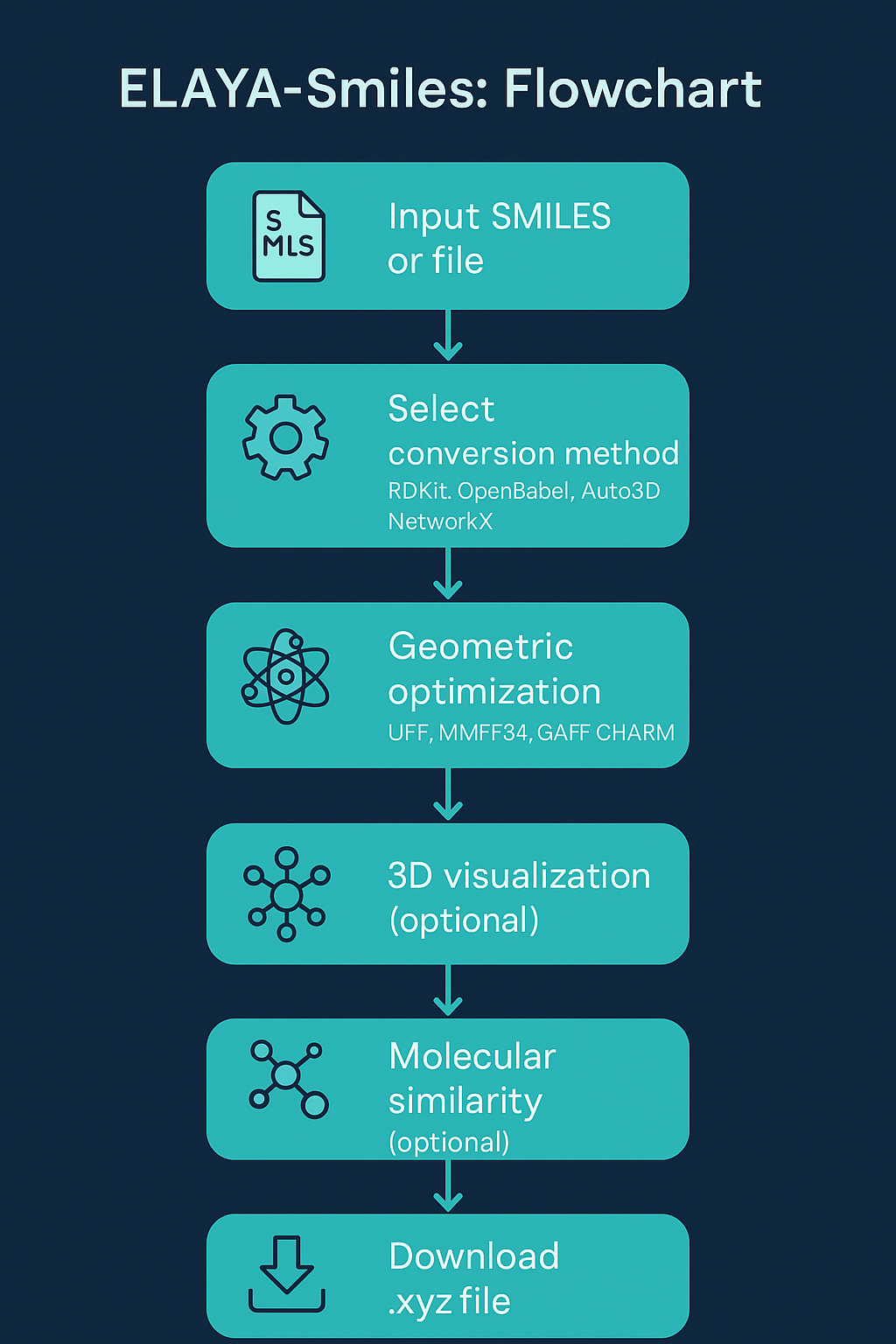
From string to structure in five steps
Enter a SMILES string or upload a batch file with one SMILES per line.
Select an algorithm (RDKit, OpenBabel, or NetworkX) and an optional force field.
Optionally run GLOMOS to sample low-energy conformers with ANI potentials.
Inspect the interactive 3D structure or switch to a 2D diagram at any time.
Download in XYZ, SDF, MOL, or MOL2 format for use in external software.
See it in action
Live conversion of a SMILES string into a fully optimized 3D structure
Technologies
| Backend | Flask · Python 3 |
| Cheminformatics | RDKit · OpenBabel · DScribe |
| Neural potentials | ANI-1ccx / ANI-2x via TorchANI |
| Visualization | 3Dmol.js · Chart.js |
| Frontend | HTML5 · CSS3 · Vanilla JavaScript |
Project Authors
ELAYA SMILES was designed and developed by Gabriela Vidales, with scientific guidance from MSc Luis Ortiz, PhD Filiberto Ortiz Chi and PhD Gabriel Merino.
Passion for building tools that bring science closer to people. Fostering open and reproducible science.
View on GitHub ↗Share your feedback
Found a bug? Have a suggestion? Something you'd like to see?
Your input helps make ELAYA better.
Committed to using science, education, and technology to improve people's quality of life.
LinkedIn ↗ GitHub ↗Bug report
Found something unexpected? Open an issue with your SMILES string and browser details.
Open an issue ↗Suggestion
Have an idea for a new feature, algorithm, or output format? I'd love to hear it.
Share an idea ↗